
Real world bioequivalence of prolonged release formulations of tacrolimus in renal transplant recipients. Is a determination needed before switching among them?
antonio franco esteve1,3, Iván Beltrá-Picó2,3, Patricio Más-Serrano2,3,4, ELena De la Cruz1,3, Isabel Gascón-Ros2, Amelia Ramón-López3,4, Ricardo Nalda-Molina3,4, Francisco Javier Pérez Contreras1,3.
1Nephrology, Hospital Dr Balmis Alicante, alicante, Spain; 2Pharmacy, Hospital Dr. Balmis Alicante, Alicante, Spain; 3Alicante Institute for Health and Biomedical Research (ISABIAL), Hospital Dr. Balmis Alicante, Alicante, Spain; 4 Division of Pharmacy and Pharmaceutics, School of Pharmacy, Miguel Hernández University, Alicante, Spain
The regulatory bioequivalence studies are conducted in healthy adults and with a single-dose. Associated pathology, repited doses and polypharmacy may affect the pharmacokinetics of these products.
The objective of this study is to determine whether the prolonged-release capsule formulation of tacrolimus, Conferoport®, is bioequivalent to Advagraf® and Tacforius® in real world,according to the paramethers of EMA and FDA :maximun concentration(Cmax)(0,8- 1,2) y area under curve (AUC).(0,9- 1,10 EMA)(0.8-1.2 FDA)
We designed 2 prospective observational studies with 2 cohorts in each study. In the first study, recipients with Advagraf(R) were switched to Conferoport and in the second from Conferoport to Tacforius.
We enroled first renal transplant recipients over 18 years from deceased donors transplanted at least two years in advance. Patients had stable renal function with tacrolimus without change in dose the last six months.They had no donor specific antibodies nor gastrointestinal disorders. All recipients received a second immunosuppressant,micophenolate mofetil or sirolimus, without prednisone.
The pharmacokinetic parameters of each cohort were studied separately and were carried out in the two periods evaluated: Period 1(treated with the reference drug) and Period 2 (after conversion to the other drug).
Patients received the same once-daily dose of both formulations in each of the two studies, with a time of 14 days between them.Drug administration was on an empty stomach. Samples were taken in troughs ( prior to the dose) and at 2 and 3 hours after administration.
Tacrolimus concentration was determined in blood by enzyme immunoassay (Dimension®, Siemens) (range: 1.2-30 ng/ml).
On each cohort, the end points were the pharmacokinetic parameter AUC from time zero to the trough concentration before next dose and Cmax from administration of study drug. Pharmacokinetic AUCs were calculated using the sampling time points and a logarithmic trapezoidal method.
A total of 41 patients were enroled(study 1 n=21, study 2 n=20), the average time since receiving transplant was 81.1 months. The mean dose of tacrolimus was 2.4 mg/24h. Baseline demographics, renal function and tacrolimus concentrations are showed.
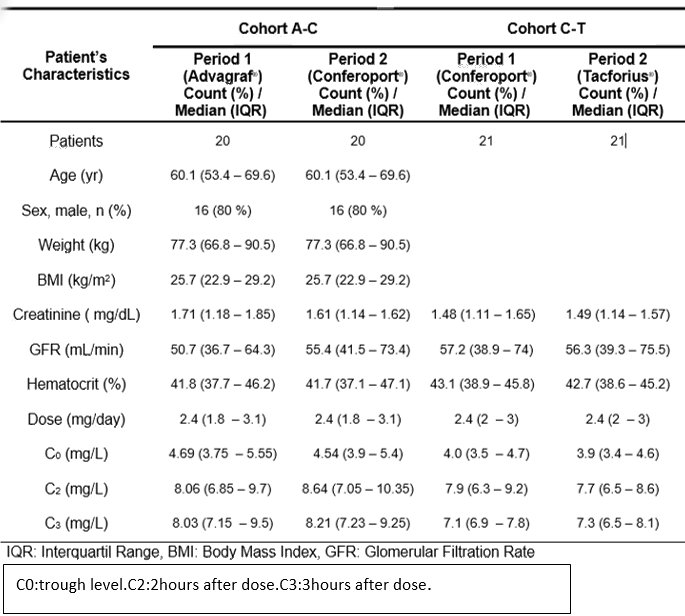
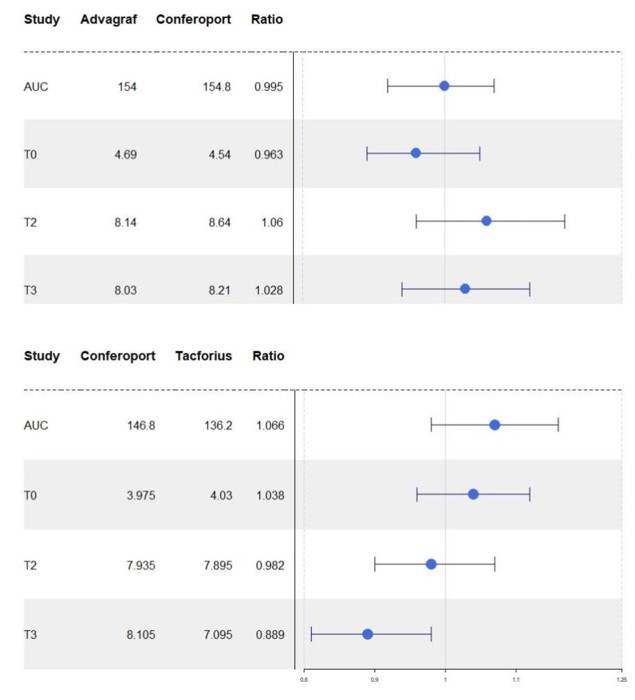
The Advagraft/Conferoport and Conferoport/Tacforius ratios of the parameters to assess bioequivalence (AUC and Cmax) were within the ratio required by the EMA and FDA .They are shown .
No side effects were observed during the study.
We confirm that in stable renal transplant recipients in the real-world, the extended-release Conferoport® is bioequivalent to the Advagraf® and Tacforius.Then , all formulations can be swithed among them without performing trough concentrations.
[1] renal transplant
[2] bioequivalence
[3] prolonged released tacrolimus
