Targeting islet inflammation by RIPK3 phosphorylation for type 2 diabetes
Jianmin Wu1, Wei Wang1, Xiaoqian Ma1.
1Cell Transplantation and Gene Therapy Institute, The Third Xiangya Hospital, Central South University, changsha, People's Republic of China, Changsha, People's Republic of China
Introduction: Inflammation plays a crucial role in the progression of type 2 diabetes (T2D). While some drugs under development aim to alleviate inflammatory damage by suppressing β cells death and dysfunction, the limited clinical application persists due to an incomplete understanding of the mechanisms underlying inflammatory injury. Here, we uncover that RIPK3 initiates cell necrosis pathways, mediating inflammatory injury in T2D. Simultaneously, we discover itaconate, a byproduct of the tricarboxylic acid cycle, which can inhibit RIPK3-mediated inflammatory damage through alkylating modification of RIPK3, ultimately slowing the progression of type 2 diabetes.
Method: These 6-week-old male C57BL/6 mice, either wild type or IRG1 KO which were itaconate-deficient, were used to establish a type 2 diabetes mellitus (T2D) model. This was achieved through a protocol involving a 4-week HFD diet (with 60% of caloric intake from fat), followed by injections of streptozotocin at a dose of 30 mg/kg three times per week. Throughout the modeling process, mice were either concurrently subjected to or exempted from intraperitoneal injections of itaconate (30 mg/kg) thrice weekly. Blood glucose levels of all mice were measured every two days, providing a basis for analyzing the impact of itaconate on the progression of T2D. Pancreases from mice under different treatments were isolated for RNA sequencing, insulin immunohistochemical staining, and Western blot experiments to assess genetic and protein changes. Mouse serum was extracted for flow cytometry analysis to evaluate the expression of inflammatory factors by cytokine beads array. The interaction between itaconate and the RIPK3 protein was examined through mass spectrometry analysis.
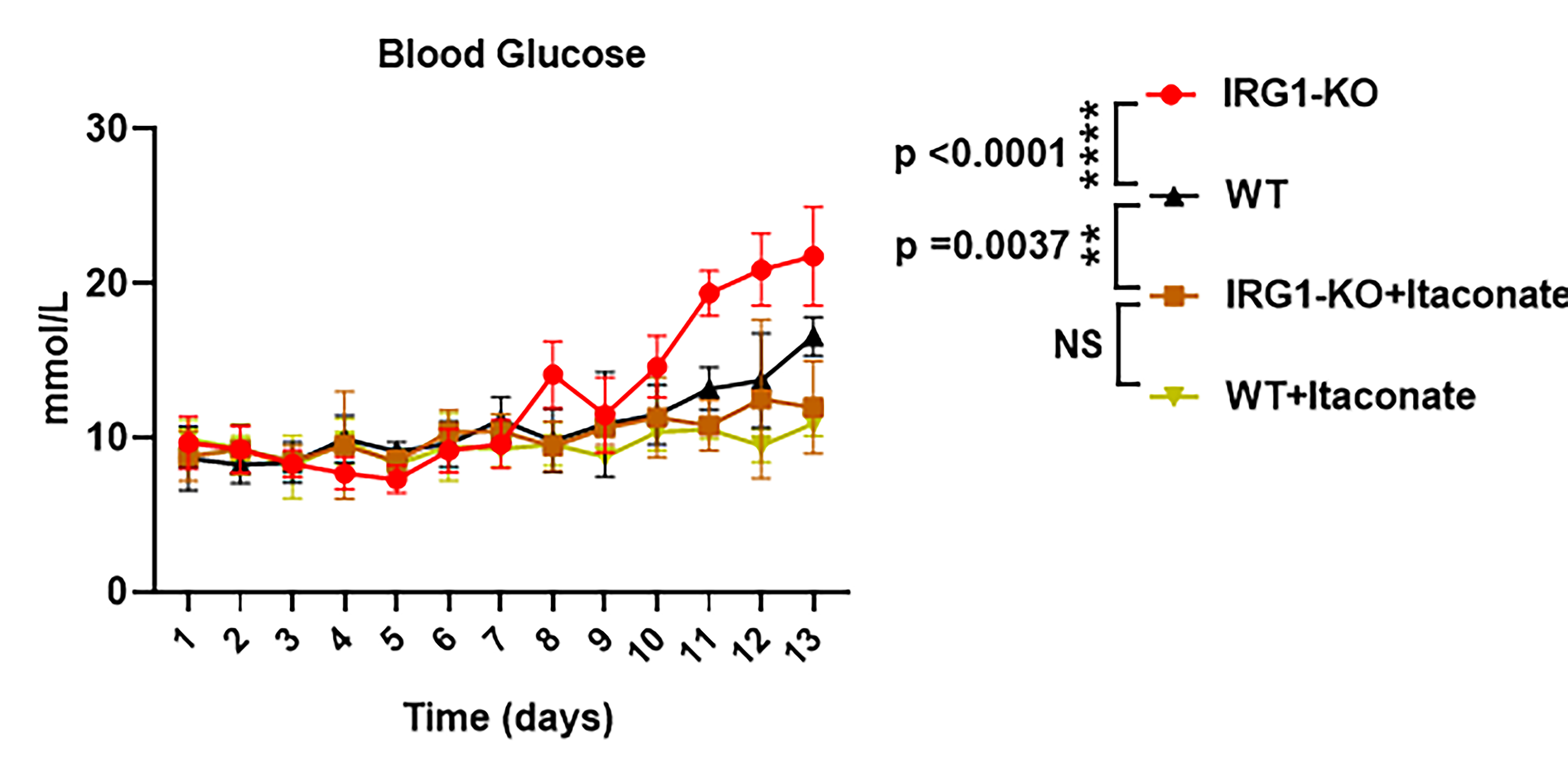
Results: In this study, we observed an aberrant downregulation of itaconate in mice of T2D. IRG1 KO mice which were itaconate-deficient exhibited increased susceptibility to diabetes and demonstrated a higher rate of blood glucose compared to wild type mice (WT:20.20±1.49 mmol/L VS IRG1-KO:17.45±1.36 mmol/L). However, mice supplemented with itaconate can reduce the increased blood glucose (WT+ Itaconate:11.36±1.63 mmol/L VS IRG1-KO+Itaconate:10.7±0.84 mmol/L). This supplementation limited immune cell infiltration and the secretion of inflammatory cytokines by inhibiting inflammatory injury.", so as to slow down the development of diabetes. Further investigation revealed that the protective effect of itaconate on islet inflammation damage is mediated through the alkylation of the C360 site on RIPK3, inhibiting phosphorylation at T231/S232. Itaconate-mediated alkylation modification of RIPK3 inhibits necroptosis pathway activation, thereby enhancing pancreatic cell viability under inflammatory injury, improving insulin secretion function, and ultimately ameliorating T2DM in mice.
Conclusion: our study suggests that itaconate may serve as a potential therapeutic target to alleviate β cell injury by inhibiting islet inflammation associated with the progression of T2D
National Natural Science Foundation of China (Grant No: 82272102).
[1] Inflammation
[2] Diabetes
[3] Allograft Survival
