Computational immuno-profiling and prediction of antibody-mediated rejection in kidney transplant recipients
Satoru Kawakita1, Vadim Jucaud1, Zhaohui Wang1, Xiling Shen1, Chongming Jiang1.
1Terasaki Institute for Biomedical Innovation, Los Angeles, CA, United States
Introduction: Antibody-mediated rejection (ABMR) is a significant obstacle to achieving long-term graft survival among kidney transplant (KT) recipients. Although the serological presence of donor-specific antibody (DSA) often accompanies ABMR, an accurate diagnosis requires more than the presence of DSA alone. Timely and precise diagnosis of ABMR remains a clinical challenge, compounded by potential inter-observer variability in the histological grading of biopsies. To address this issue, we investigated whether (1) unique computationally-derived immune-cell signatures are associated with ABMR and (2) machine learning (ML) models trained with these features can reliably and systematically distinguish non-ABMR from ABMR patients.
Method: Microarray data (GSE50084) was obtained for tissue biopsy samples from KT recipients collected at the time of clinically-indicated biopsy. Patients were categorized into three groups: without DSA (DSA(-); n = 20), with DSA and no ABMR (DSA(+)/ABMR(-); n = 13), and with DSA and ABMR (DSA(+)/ABMR(+); n = 28). We performed immune cell infiltration analysis using the quanTIseq method to calculate the fractions of 10 immune cell types and statistically compared them between patient groups. We then trained XGBoost models with n-repeated K-fold cross-validation using selected features to classify ABMR vs. non-ABMR patients.
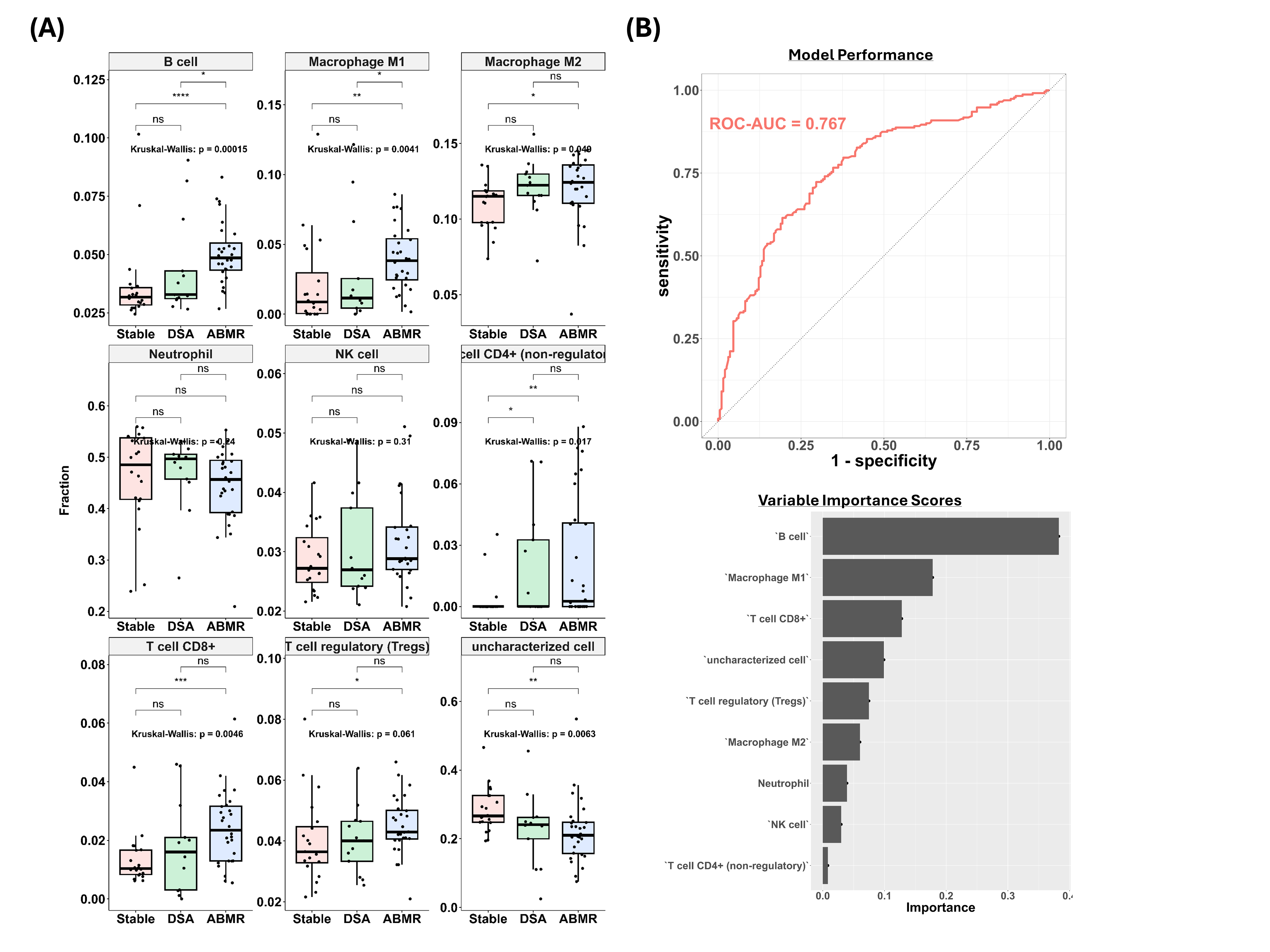
Results: Immune cell infiltration analysis revealed that compared to the DSA(-) control, B cells, macrophages, and a subset of T cells were upregulated in the ABMR group (Figure A). Furthermore, the differentiating factors between DSA(+)/ABMR(-) and DSA(+)/ABMR(+) groups were B cells and macrophage M1. When XGBoost models were trained on selected immune-cell types, the best-performing model exhibited an area under the receiver operating characteristic curve of 0.767. The model also calculated variable importance (VI) scores for input features, identifying B cells (VI = 0.352) as the most significant feature, followed by macrophage M1 (VI = 0.192) and CD8+ T cells (VI = 0.131).
Conclusion: Computational immunophenotyping of biopsy samples revealed distinct immune-cell profiles for the three patient groups, differentiating ABMR group from non-ABMR group with or without DSA. When ML models were trained with these computationally-derived features, they exhibited good model performance despite the relatively small size of the training set. This study demonstrates the potential of coupling ML with computational immuno-profiling to develop a cost-efficient and effective model for the systematic diagnosis of ABMR. Future efforts should focus on further training and validating the ML model with more data and improving model parsimony. The practical utility of such computational models may be enhanced by using blood samples instead of biopsy samples for non-invasive clinical diagnostics or prognostics.
We thank Drs. Pilib Ó Broin and Enver Akalin for depositing the data (GSE50084) to the Gene Expression Omnibus repository and making it accessible to the research community.
[1] Antibody-mediated Rejection
[2] Machine Learning
[3] Bioinformatics
